| Autosomal dominant polycystic kidney disease | |
|---|---|
| Other names | Autosomal dominant PKD, adult-onset PKD |
 | |
| Polycystic kidneys | |
| Specialty | Medical genetics |
Autosomal dominant polycystic kidney disease (ADPKD) is one of the most common, life-threatening inherited human disorders and the most common hereditary kidney disease. It is associated with large interfamilial and intrafamilial variability, which can be explained to a large extent by its genetic heterogeneity and modifier genes. It is also the most common of the inherited cystic kidney diseases — a group of disorders with related but distinct pathogenesis, characterized by the development of renal cysts and various extrarenal manifestations, which in case of ADPKD include cysts in other organs, such as the liver, seminal vesicles, pancreas, and arachnoid membrane, as well as other abnormalities, such as intracranial aneurysms and dolichoectasias, aortic root dilatation and aneurysms, mitral valve prolapse, and abdominal wall hernias. Over 50% of patients with ADPKD eventually develop end stage kidney disease and require dialysis or kidney transplantation. ADPKD is estimated to affect at least one in every 1000 individuals worldwide, making this disease the most common inherited kidney disorder with a diagnosed prevalence of 1:2000 and incidence of 1:3000-1:8000 in a global scale.
Signs and symptoms
Among the clinical presentation are:
- Acute loin pain
- Blood in the urine
- Ballotable kidneys
- Subarachnoid hemorrhage (berry aneurysm)
- Hypertension
- Associated liver cysts
- Uremia due to kidney failure
- Anemia due to chronic kidney disease
- Increase RBC or erythropoietin secretion
Signs and symptoms of ADPKD often develop between 30 and 40 years of age.
Genetics
ADPKD is genetically heterogeneous with two genes identified: PKD1 (chromosome region 16p13.3; around 85% cases) and PKD2 (4q21; around 15% cases). Several genetic mechanisms probably contribute to the phenotypic expression of the disease. Although evidence exists for a two-hit mechanism (germline and somatic inactivation of two PKD alleles) explaining the focal development of renal and hepatic cysts, haploinsufficiency is more likely to account for the vascular manifestations of the disease. Additionally, new mouse models homozygous for PKD1 hypomorphic alleles 22 and 23 and the demonstration of increased renal epithelial cell proliferation in PKD2 +/− mice suggest that mechanisms other than the two-hit hypothesis also contribute to the cystic phenotype.
Large interfamilial and intrafamilial variability occurs in ADPKD. Most individuals with PKD1 mutations have kidney failure by age 70 years, whereas more than 50% of individuals with PKD2 mutations have adequate renal function at that age (mean age of onset of end-stage renal disease: 54·3 years with PKD1; 74·0 years with PKD2).
The significant intrafamilial variability observed in the severity of renal and extrarenal manifestations points to genetic and environmental modifying factors that may influence the outcome of ADPKD, and results of an analysis of the variability in renal function between monozygotic twins and siblings support the role of genetic modifiers in this disease. It is estimated that 43–78% of the variance in age to ESRD could be due to heritable modifying factors, with parents as likely as children to show more severe disease in studies of parent-child pairs.
Pathophysiology
In many patients with ADPKD, kidney dysfunction is not clinically apparent until 30 or 40 years of life. However, an increasing body of evidence suggests the formation of renal cysts starts in utero. Cysts initially form as small dilations in renal tubules, which then expand to form fluid-filled cavities of different sizes. Factors suggested to lead to cystogenesis include a germline mutation in one of the polycystin gene alleles, a somatic second hit that leads to the loss of the normal allele, and a third hit, which can be a renal insult that triggers cell proliferation, and an injury response. Due to numerous similarities between the pathophysiology of ADPKD and the pathophysiology of the renal response to injury, ADPKD has been described as a state of aberrant and persistent activation of renal injury response pathways. In the progression of the disease, continued dilation of the tubules through increased cell proliferation, fluid secretion, and separation from the parental tubule lead to the formation of cysts.
ADPKD, together with many other diseases that present with renal cysts, can be classified into a family of diseases known as ciliopathies. Epithelial cells of the renal tubules, including all the segments of the nephron and the collecting ducts (with the exception of intercalated cells) show the presence of a single primary apical cilium. Polycystin-1, the protein encoded by the PKD1 gene, is present on these cilia and is thought to sense the flow with its large extracellular domains, activating the calcium channels associated with polycystin-2, the product of gene PKD2, as a result of the genetic setting of ADPKD as explained in the genetics sub-section above.
Epithelial cell proliferation and fluid secretion that lead to cystogenesis are two hallmark features in ADPKD. During the early stages of cystogenesis, cysts are attached to their parental renal tubules and a derivative of the glomerular filtrate enters the cysts. Once these cysts expand to approximately 2 mm in diameter, the cyst closes off from its parental tubule and after that fluid can only enter the cysts through transepithelial secretion, which in turn is suggested to increase due to secondary effects from an increased intracellular concentration of cyclic AMP (cAMP).
Clinically, the insidious increase in the number and size of renal cysts translates as a progressive increment in kidney volume. Studies led by Mayo Clinic professionals established that the total kidney volume (TKV) in a large cohort of ADPKD patients was 1060 ± 642ml with a mean increase of 204ml over three years, or 5.27% per year in the natural course of the disease, among other important, novel findings that were extensively studied for the first time.

Diagnosis
Usually, the diagnosis of ADPKD is initially performed by renal imaging using ultrasound, CT scan, or MRI. However, molecular diagnostics can be necessary in the following situations: 1- when a definite diagnosis is required in young individuals, such as a potential living related donor in an affected family with equivocal imaging data; 2- in patients with a negative family history of ADPKD, because of potential phenotypic overlap with several other kidney cystic diseases; 3- in families affected by early-onset polycystic kidney disease, since in this cases hypomorphic alleles and/or oligogenic inheritance can be involved; and 4- in patients requesting genetic counseling, especially in couples wishing a pre-implantation genetic diagnosis.
The findings of large echogenic kidneys without distinct macroscopic cysts in an infant/child at 50% risk for ADPKD are diagnostic. In the absence of a family history of ADPKD, the presence of bilateral renal enlargement and cysts, with or without the presence of hepatic cysts, and the absence of other manifestations suggestive of a different renal cystic disease provide presumptively, but not definite, evidence for the diagnosis. In some cases, intracranial aneurysms can be an associated sign of ADPKD, and screening can be recommended for patients with a family history of intracranial aneurysm.
Molecular genetic testing by linkage analysis or direct mutation screening is clinically available; however, genetic heterogeneity is a significant complication to molecular genetic testing. Sometimes, a relatively large number of affected family members need to be tested in order to establish which one of the two possible genes is responsible within each family. The large size and complexity of PKD1 and PKD2 genes, as well as marked allelic heterogeneity, present obstacles to molecular testing by direct DNA analysis. The sensitivity of testing is nearly 100% for all patients with ADPKD who are age 30 years or older and for younger patients with PKD1 mutations; these criteria are only 67% sensitive for patients with PKD2 mutations who are younger than age 30.
-
 Adult polycystic kidney
Adult polycystic kidney
-
 Diagram of autosomal dominant polycystic disease with a normal kidney inset for comparison
Diagram of autosomal dominant polycystic disease with a normal kidney inset for comparison
-
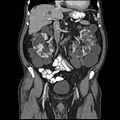 Abdominal CT scan of an adult with autosomal dominant polycystic kidney disease: Extensive cyst formation is seen over both kidneys, with a few cysts in the liver, as well. (Coronal plane)
Abdominal CT scan of an adult with autosomal dominant polycystic kidney disease: Extensive cyst formation is seen over both kidneys, with a few cysts in the liver, as well. (Coronal plane)



Treatment
Currently, the only pharmacological treatment available for ADPKD consists in reducing the speed in gain of total kidney volume (TKV) with vasopressin receptor 2 (V2) antagonists (i.e. tolvaptan). Tolvaptan treatment does not halt or reverse disease progression and patients still progress towards renal failure. Palliative treatment modalities involve symptomatic medications (nonopioid and opioid analgesics) for abdominal/retroperitoneal pain. Options for analgesic-resistant pain include simple or complex surgical procedures (i.e. renal cyst aspiration, cyst decortication, renal denervation and nephrectomy), which can result in complications inherent to surgery. Recent research suggests that ketogenic dietary interventions beneficially affect the progression and symptoms in individuals with ADPKD. Mild weight loss favorably affects pain indicating the benefit of dietary and lifestyle changes.
Aquaretic medication
In 2014, Japan was the first country in the world to approve a pharmacological treatment for ADPKD followed by Canada and Europe, which approved the drug tolvaptan for ADPKD patients in the beginning of 2015. The USA FDA approved the use of tolvaptan in the treatment of ADPKD in 2018. Tolvaptan, an aquaretic drug, is a vasopressin receptor 2 (V2) antagonist. Pre-clinical studies had suggested that the molecule cAMP could be involved in the enlargement of ADPKD cysts, and studies on rodents confirmed the role of vasopressin in increasing the levels of cAMP in the kidney, which laid the basis for the conduction of clinical studies. Because data from the Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) led by Mayo Clinic showed that total kidney volume (TKV) predicted the risk of developing chronic kidney disease in patients with ADPKD, the TEMPO 3:4 trial, which enrolled patients from 129 sites worldwide from 2007 to 2009, evaluated TKV as a primary end-point to test the efficacy of tolvaptan in ADPKD patients. That study showed a significant decrease in the ratio of TKV increase and deterring of renal function decline in ADPKD patients after treatment with tolvaptan; however, because laboratory test results regarding liver function appeared elevated in a percentage of patients enrolled in that study, the approval of the drug was either delayed by regulatory agencies or, as in case of the US, altogether denied.
Dietary and lifestyle interventions
Research using ADPKD mouse models showed that mild food restriction strongly improved disease progression. The mechanism was shown to involve the metabolic state of ketosis, and beneficial effects could be produced by time-restricted feeding, acute fasting, a ketogenic diet, or by supplementation with the ketone beta-hydroxybutyrate in mouse, rat and cat models of ADPKD. A ketogenic diet regimen not only halted further disease progression but led to partial reversal of renal cystic disease in a rat model. The metabolic state of ketosis may be beneficial in ADPKD because renal cyst cells in ADPKD have a metabolic defect similar to the Warburg effect in cancer that makes them highly dependent on glucose, and unable to metabolize fatty acids and ketones. Consistent with this, serum glucose levels positively correlate with faster disease progression in ADPKD patients. Also, individuals with ADPKD and type 2 diabetes have significantly larger total kidney volume (TKV) than those with ADPKD alone, and overweight or obesity associate with faster progression in early-stage ADPKD. A retrospective case series study showed that ADPKD disease symptoms - including pain, hypertension and renal function - improved among 131 patients who implemented ketogenic diets for an average duration of 6 months.
Dietary intake of sodium is associated with worse renal function decline in ADPKD, and limiting sodium intake is generally recommended to patients. Dietary protein intake was not found to correlate with ADPKD progression.
Increased water intake is thought to be beneficial in ADPKD and is generally recommended. The underlying beneficial mechanism of increased water intake may be related to effects on the vasopressin V2 receptor or may be due to the suppression of harmful micro-crystal formation in renal tubules by dilution of solutes such as calcium oxalate, calcium phosphate and uric acid.
Dietary intake of oxalate or inorganic phosphate has been shown to accelerate PKD disease progression in several rodent models. Low levels or urinary citrate – a natural antagonist of the formation of harmful crystals in kidney tubules – have been shown to associate with worse disease progression in ADPKD patients.
Analgesic medication
Chronic pain in patients with ADPKD is often refractory to conservative, noninvasive treatments, but nonopioid analgesics and conservative interventions can be first used before opioid analgesics are considered; if pain continues, then surgical interventions can target renal or hepatic cysts to directly address the cause of pain, with surgical options including renal cyst decortication, renal denervation, and nephrectomy.
Renal cyst aspiration
Aspiration with ethanol sclerotherapy can be performed for the treatment of symptomatic simple renal cysts, but can be impractical in advanced patients with multiple cysts. The procedure itself consists in the percutaneous insertion of a needle into the identified cyst, under ultrasound guidance, with subsequent draining the contained liquid; the sclerotherapy is used to avoid liquid reaccumulation that can occur in the cyst, which can result in symptom recurrence.
Laparoscopic cyst decortication
Laparoscopic cyst decortication (also referred to as marsupialization) consists in the removal of one or more kidney cysts through laparoscopic surgery, during which cysts are punctured, and the outer wall of the larger cysts is excised with care not to incise the renal parenchyma. This procedure can be useful for pain relief in patients with ADPKD, and is usually indicated after earlier cyst aspiration has confirmed that the cyst to be decorticated is responsible for pain. Nonrandomised controlled trials conducted in the '90s showed that patients with symptomatic simple renal cysts who had recurrence of symptoms after initial response to simple aspiration could be safely submitted to cyst decortication, with a mean pain-free life between 17 and 24 months after surgery. Laparoscopic decortication presents a 5% recurrence rate of renal cysts compared to an 82% recurrence rate obtained with sclerotherapy.
Neurolysis
A novel treatment of specifically the chronic pain experienced by many with ADPKD is Celiac plexus neurolysis. This involves the chemical ablation of the celiac plexus, to cause a temporary degeneration of targeted nerve fibers. When the nerve fibers degenerate, it causes an interruption in the transmission of nerve signals. This treatment, when successful, provides significant pain relief for a period ranging from a few days to over a year. The procedure may be repeated when the affected nerves have healed and the pain returns.
Nephrectomy
Many ADPKD patients experience symptomatic sequelae in consequence of the disease, such as cyst hemorrhage, flank pain, recurrent infections, nephrolithiasis, and symptoms of mass effect (i.e., early satiety, nausea and vomiting, and abdominal discomfort), from their enlarged kidneys. In such cases, nephrectomy can be required due to intractable symptoms or when in the course of preparing for kidney transplantation, the native kidneys are found to impinge upon the true pelvis and preclude the placement of a donor allograft. Additionally, native nephrectomy may be undertaken in the presence of suspected malignancy, as renal cell carcinoma (RCC) is two to three times more likely in the ADPKD population in end-stage kidney disease (ESKD) than in the ESKD patients without ADPKD. Although the indications for nephrectomy in ADPKD may be related to kidney size, the decision to proceed with native nephrectomy is often undertaken on an individual basis, without specific reference to kidney size measurements.
Dialysis
Two modalities of dialysis can be used in the treatment of ADPKD patients: peritoneal dialysis and hemodialysis. Epidemiological data shows that ADPKD affects 5–13.4% of patients undergoing hemodialysis in Europe and in the United States, and about 3% in Japan. Peritoneal dialysis has usually been contra-indicated in ADPKD patients with large kidney and liver volumes, due to expected physical difficulties in the procedure and possible complications; however, no difference is seen in long-term morbidity between hemodialysis and peritoneal dialysis in ADPKD.
Kidney transplant
Kidney transplantation is accepted as the preferred treatment for ADPKD patients with ESRD. Among American patients on the kidney-transplant waiting list (as of December 2011), 7256 (8.4%) were listed due to cystic kidney disease and of the 16,055 renal transplants performed in 2011, 2057 (12.8%) were done for patients with cystic kidney disease, with 1,189 from deceased donors and 868 from living donors.
Prognosis
In ADPKD patients, gradual cyst development and expansion result in kidney enlargement, and during the course of the disease, glomerular filtration rate remains normal for decades before kidney function starts to progressively deteriorate, making early prediction of renal outcome difficult. The CRISP study, mentioned in the treatment section above, contributed to build a strong rationale supporting the prognostic value of total kidney volume (TKV) in ADPKD; TKV (evaluated by MRI) increases steadily and a higher rate of kidney enlargement correlated with accelerated decline of GFR, while patient height-adjusted TKV (HtTKV) ≥600 ml/m predicts the development of stage 3 chronic kidney disease within 8 years.
Besides TKV and HtTKV, the estimated glomerular filtration rate (eGFR) has also been tentatively used to predict the progression of ADPKD. After the analysis of CT or MRI scans of 590 patients with ADPKD treated at the Mayo Translational Polycystic Kidney Disease Center, Irazabal and colleagues developed an imaging-based classification system to predict the rate of eGFR decline in patients with ADPKD. In this prognostic method, patients are divided into five subclasses of estimated kidney growth rates according to age-specific HtTKV ranges (1A, <1.5%; 1B, 1.5–3.0%; 1C, 3.0–4.5%; 1D, 4.5–6.0%; and 1E, >6.0%) as delineated in the CRISP study. The decline in eGFR over the years following initial TKV measurement is significantly different between all five patient subclasses, with those in subclass 1E having the most rapid decline. Some of the most common causes of death in patients with ADPKD are various infections (25%), a ruptured berry aneurysm (15%), or coronary/hypertensive heart disease (40%).
References
- ^ Torres VE, Harris PC, Pirson Y (April 2007). "Autosomal dominant polycystic kidney disease". Lancet. 369 (9569): 1287–1301. doi:10.1016/S0140-6736(07)60601-1. PMID 17434405. S2CID 1700992.
- "What is ADPKD?". PKD Foundation. Retrieved 2022-09-23.
- Dalgaard OZ (1957). "Bilateral polycystic disease of the kidneys; a follow-up of two hundred and eighty-four patients and their families". Acta Medica Scandinavica. Supplementum. 328: 1–255. PMID 13469269.
- Torres VE, Harris PC (July 2009). "Autosomal dominant polycystic kidney disease: the last 3 years". Kidney International. 76 (2): 149–168. doi:10.1038/ki.2009.128. PMC 2812475. PMID 19455193.
- ^ Grantham JJ (October 2008). "Clinical practice. Autosomal dominant polycystic kidney disease". The New England Journal of Medicine. 359 (14): 1477–1485. doi:10.1056/NEJMcp0804458. PMC 2843931. PMID 18832246.; Reprinted in Niemczyk M, Niemczyk S, Paczek L (2009). "Autosomal dominant polycystic kidney disease and transplantation". Annals of Transplantation. 14 (4): 86–90. PMC 2843931. PMID 20009161.
- Muto S, Kawano H, Higashihara E, Narita I, Ubara Y, Matsuzaki T, et al. (October 2015). "The effect of tolvaptan on autosomal dominant polycystic kidney disease patients: a subgroup analysis of the Japanese patient subset from TEMPO 3:4 trial". Clinical and Experimental Nephrology. 19 (5): 867–877. doi:10.1007/s10157-015-1086-2. PMID 25663351. S2CID 12124902.
- ^ Higashihara E, Nutahara K, Kojima M, Tamakoshi A, Yoshiyuki O, Sakai H, Kurokawa K (December 1998). "Prevalence and renal prognosis of diagnosed autosomal dominant polycystic kidney disease in Japan". Nephron. 80 (4): 421–427. doi:10.1159/000045214. PMID 9832641. S2CID 22124996.
- Levy M, Feingold J (September 2000). "Estimating prevalence in single-gene kidney diseases progressing to renal failure". Kidney International. 58 (3): 925–943. doi:10.1046/j.1523-1755.2000.00250.x. PMID 10972657.
- ^ Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Grantham JJ, Higashihara E, et al. (December 2012). "Tolvaptan in patients with autosomal dominant polycystic kidney disease". The New England Journal of Medicine. 367 (25): 2407–2418. doi:10.1056/NEJMoa1205511. PMC 3760207. PMID 23121377.
- ^ Cornec-Le Gall E, Le Meur Y (November 2014). "". Nephrologie & Therapeutique. 10 (6): 433–440. doi:10.1016/j.nephro.2014.03.003. PMID 25086476.
- "Polycystic kidney disease". Mayo Clinic. Retrieved 2022-05-23.
- Somlo, Stefan; Wirth, Brunhilde; Germino, Gregory G.; Weinstat-Saslow, Debra; Gillespie, Gerald A. J.; Himmelbauer, Heinz; Steevens, Laura; Coucke, Paul; Willems, Patrick; Bachner, Lucien; Coto, Eliecer; López-Larrea, Carlos; Peral, Bélen; Millán, JoséLuis San; Saris, Jasper J. (1992-05-01). "Fine genetic localization of the gene for autosomal dominant polycystic kidney disease (PKD1) with respect to physically mapped markers". Genomics. 13 (1): 152–158. doi:10.1016/0888-7543(92)90215-E. ISSN 0888-7543.
- Kimberling, William J.; Kumar, Shrawan; Gabow, Patricia A.; Kenyon, Judith B.; Connolly, Christopher J.; Somlo, Stefan (1993-12-01). "Autosomal dominant polycystic kidney disease: Localization of the second gene to chromosome 4q13–q23". Genomics. 18 (3): 467–472. doi:10.1016/S0888-7543(11)80001-7. ISSN 0888-7543.
- Kumar, Shrawan; Kimberling, William J.; Gabow, Patricia A.; Kenyon, Judy B. (1991-06-01). "Genetic linkage studies of autosomal dominant polycystic kidney disease: search for the second gene in a large Sicilian family". Human Genetics. 87 (2): 129–133. doi:10.1007/BF00204167. ISSN 1432-1203.
- Torra R, Badenas C, San Millán JL, Pérez-Oller L, Estivill X, Darnell A (August 1999). "A loss-of-function model for cystogenesis in human autosomal dominant polycystic kidney disease type 2". American Journal of Human Genetics. 65 (2): 345–352. doi:10.1086/302501. PMC 1377933. PMID 10417277.
- Watnick TJ, Torres VE, Gandolph MA, Qian F, Onuchic LF, Klinger KW, et al. (August 1998). "Somatic mutation in individual liver cysts supports a two-hit model of cystogenesis in autosomal dominant polycystic kidney disease". Molecular Cell. 2 (2): 247–251. doi:10.1016/s1097-2765(00)80135-5. PMID 9734362.
- Qian Q, Hunter LW, Li M, Marin-Padilla M, Prakash YS, Somlo S, et al. (August 2003). "Pkd2 haploinsufficiency alters intracellular calcium regulation in vascular smooth muscle cells". Human Molecular Genetics. 12 (15): 1875–1880. doi:10.1093/hmg/ddg190. PMID 12874107.
- Gao Z, Joseph E, Ruden DM, Lu X (April 2004). "Drosophila Pkd2 is haploid-insufficient for mediating optimal smooth muscle contractility". The Journal of Biological Chemistry. 279 (14): 14225–14231. doi:10.1074/jbc.M312223200. PMID 14732716.
- Hateboer N, v Dijk MA, Bogdanova N, Coto E, Saggar-Malik AK, San Millan JL, et al. (January 1999). "Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 Study Group". Lancet. 353 (9147): 103–107. doi:10.1016/s0140-6736(98)03495-3. PMID 10023895. S2CID 30757096.
- Persu A, Duyme M, Pirson Y, Lens XM, Messiaen T, Breuning MH, et al. (December 2004). "Comparison between siblings and twins supports a role for modifier genes in ADPKD". Kidney International. 66 (6): 2132–2136. doi:10.1111/j.1523-1755.2004.66003.x. PMID 15569302.
- Fain PR, McFann KK, Taylor MR, Tison M, Johnson AM, Reed B, Schrier RW (April 2005). "Modifier genes play a significant role in the phenotypic expression of PKD1". Kidney International. 67 (4): 1256–1267. doi:10.1111/j.1523-1755.2005.00203.x. PMID 15780078.
- Paterson AD, Magistroni R, He N, Wang K, Johnson A, Fain PR, et al. (March 2005). "Progressive loss of renal function is an age-dependent heritable trait in type 1 autosomal dominant polycystic kidney disease". Journal of the American Society of Nephrology. 16 (3): 755–762. doi:10.1681/ASN.2004090758. PMID 15677307.
- Geberth S, Ritz E, Zeier M, Stier E (1995). "Anticipation of age at renal death in autosomal dominant polycystic kidney disease (ADPKD)?". Nephrology, Dialysis, Transplantation. 10 (9): 1603–1606. PMID 8559477.
- ^ Paul BM, Vanden Heuvel GB (2014). "Kidney: polycystic kidney disease". Wiley Interdisciplinary Reviews. Developmental Biology. 3 (6): 465–487. doi:10.1002/wdev.152. PMC 4423807. PMID 25186187.
- Weimbs T (May 2011). "Third-hit signaling in renal cyst formation". Journal of the American Society of Nephrology. 22 (5): 793–795. doi:10.1681/ASN.2011030284. PMC 5619655. PMID 21493772.
- Weimbs T (November 2007). "Polycystic kidney disease and renal injury repair: common pathways, fluid flow, and the function of polycystin-1". American Journal of Physiology. Renal Physiology. 293 (5): F1423–F1432. doi:10.1152/ajprenal.00275.2007. PMID 17715262.
- Igarashi P, Somlo S (September 2002). "Genetics and pathogenesis of polycystic kidney disease". Journal of the American Society of Nephrology. 13 (9): 2384–2398. doi:10.1097/01.asn.0000028643.17901.42. PMID 12191984.
- Parnell SC, Magenheimer BS, Maser RL, Zien CA, Frischauf AM, Calvet JP (May 2002). "Polycystin-1 activation of c-Jun N-terminal kinase and AP-1 is mediated by heterotrimeric G proteins". The Journal of Biological Chemistry. 277 (22): 19566–19572. doi:10.1074/jbc.M201875200. PMID 11912216.
- Berbari NF, O'Connor AK, Haycraft CJ, Yoder BK (July 2009). "The primary cilium as a complex signaling center". Current Biology. 19 (13): R526–R535. doi:10.1016/j.cub.2009.05.025. PMC 2814769. PMID 19602418.
- Reed BY, McFann K, Bekheirnia MR, Reza Bekheirnia M, Nobakhthaghighi N, Nobkhthaghighi N, et al. (February 2008). "Variation in age at ESRD in autosomal dominant polycystic kidney disease". American Journal of Kidney Diseases. 51 (2): 173–183. doi:10.1053/j.ajkd.2007.10.037. PMC 2747334. PMID 18215695.
- Chapin HC, Caplan MJ (November 2010). "The cell biology of polycystic kidney disease". The Journal of Cell Biology. 191 (4): 701–710. doi:10.1083/jcb.201006173. PMC 2983067. PMID 21079243.
- Belibi FA, Reif G, Wallace DP, Yamaguchi T, Olsen L, Li H, et al. (September 2004). "Cyclic AMP promotes growth and secretion in human polycystic kidney epithelial cells". Kidney International. 66 (3): 964–973. doi:10.1111/j.1523-1755.2004.00843.x. PMID 15327388.
- ^ Torres VE (March 2010). "Treatment strategies and clinical trial design in ADPKD". Advances in Chronic Kidney Disease. 17 (2): 190–204. doi:10.1053/j.ackd.2010.01.006. PMC 4127876. PMID 20219622.
- ^ Trujillano D, Bullich G, Ossowski S, Ballarín J, Torra R, Estivill X, Ars E (September 2014). "Diagnosis of autosomal dominant polycystic kidney disease using efficient PKD1 and PKD2 targeted next-generation sequencing". Molecular Genetics & Genomic Medicine. 2 (5): 412–421. doi:10.1002/mgg3.82. PMC 4190876. PMID 25333066.
- Bergmann C, von Bothmer J, Ortiz Brüchle N, Venghaus A, Frank V, Fehrenbach H, et al. (November 2011). "Mutations in multiple PKD genes may explain early and severe polycystic kidney disease". Journal of the American Society of Nephrology. 22 (11): 2047–2056. doi:10.1681/ASN.2010101080. PMC 3279997. PMID 22034641.
- Harris PC, Rossetti S (April 2010). "Molecular diagnostics for autosomal dominant polycystic kidney disease". Nature Reviews. Nephrology. 6 (4): 197–206. doi:10.1038/nrneph.2010.18. PMC 4050432. PMID 20177400.
- Rozenfeld MN, Ansari SA, Shaibani A, Russell EJ, Mohan P, Hurley MC (January 2014). "Should patients with autosomal dominant polycystic kidney disease be screened for cerebral aneurysms?". AJNR. American Journal of Neuroradiology. 35 (1): 3–9. doi:10.3174/ajnr.A3437. PMC 7966475. PMID 23292526. S2CID 5777115.
- "Autosomal Dominant Polycystic Kidney Disease". The Lecturio Medical Concept Library. Retrieved 3 July 2021.
- ^ Strubl S, Oehm S, Torres JA, Grundmann F, Haratani J, Decker M, et al. (2021). "Ketogenic dietary interventions in autosomal dominant polycystic kidney disease—a retrospective case series study: first insights into feasibility, safety and effects". Clinical Kidney Journal. 15 (6): 1079–1092. doi:10.1093/ckj/sfab162. PMC 9155228. PMID 35664270.
- Nowak KL, Murray K, You Z, Gitomer B, Brosnahan G, Abebe KZ, et al. (July 1, 2021). "Pain and Obesity in Autosomal Dominant Polycystic Kidney Disease: A Post Hoc Analysis of the Halt Progression of Polycystic Kidney Disease (HALT-PKD) Studies". Kidney Medicine. 3 (4): 536–545.e1. doi:10.1016/j.xkme.2021.03.004. PMC 8350824. PMID 34401721.
- "Tolvaptan Cleared in US for ADPKD in Adults". 2018-04-26.
- Hanaoka K, Guggino WB (July 2000). "cAMP regulates cell proliferation and cyst formation in autosomal polycystic kidney disease cells". Journal of the American Society of Nephrology. 11 (7): 1179–1187. doi:10.1681/ASN.V1171179. PMID 10864573.
- Juul KV, Bichet DG, Nielsen S, Nørgaard JP (May 2014). "The physiological and pathophysiological functions of renal and extrarenal vasopressin V2 receptors". American Journal of Physiology. Renal Physiology. 306 (9): F931–F940. doi:10.1152/ajprenal.00604.2013. PMID 24598801.
- ^ Irazabal MV, Rangel LJ, Bergstralh EJ, Osborn SL, Harmon AJ, Sundsbak JL, et al. (January 2015). "Imaging classification of autosomal dominant polycystic kidney disease: a simple model for selecting patients for clinical trials". Journal of the American Society of Nephrology. 26 (1): 160–172. doi:10.1681/ASN.2013101138. PMC 4279733. PMID 24904092.
- Kelsey R (January 2013). "Polycystic kidney disease: Tolvaptan in ADPKD-TEMPO 3:4 trial results". Nature Reviews. Nephrology. 9 (1): 1. doi:10.1038/nrneph.2012.236. PMID 23183839. S2CID 22942772.
- Brown T (2013). "Tolvaptan Not Recommended for ADPKD". Medscape.
- Kipp KR, Rezaei M, Lin L, Dewey EC, Weimbs T (April 2016). "A mild reduction of food intake slows disease progression in an orthologous mouse model of polycystic kidney disease". American Journal of Physiology. Renal Physiology. 310 (8): F726–F731. doi:10.1152/ajprenal.00551.2015. PMC 4835927. PMID 26764208.
- ^ Torres JA, Kruger SL, Broderick C, Amarlkhagva T, Agrawal S, Dodam JR, et al. (December 2019). "Ketosis Ameliorates Renal Cyst Growth in Polycystic Kidney Disease". Cell Metabolism. 30 (6): 1007–1023.e5. doi:10.1016/j.cmet.2019.09.012. PMC 6904245. PMID 31631001.
- ^ Carney EF (January 2020). "Ketosis slows the progression of PKD". Nature Reviews. Nephrology. 16 (1): 1. doi:10.1038/s41581-019-0226-4. PMID 31654043. S2CID 204886698.
- Nowak KL, Hopp K (April 2020). "Metabolic Reprogramming in Autosomal Dominant Polycystic Kidney Disease: Evidence and Therapeutic Potential". Clinical Journal of the American Society of Nephrology. 15 (4): 577–584. doi:10.2215/CJN.13291019. PMC 7133124. PMID 32086281.
- Padovano V, Podrini C, Boletta A, Caplan MJ (November 2018). "Metabolism and mitochondria in polycystic kidney disease research and therapy". Nature Reviews. Nephrology. 14 (11): 678–687. doi:10.1038/s41581-018-0051-1. PMID 30120380. S2CID 52033674.
- Torres JA, Kruger SL, Broderick C, Amarlkhagva T, Agrawal S, Dodam JR, et al. (December 2019). "Ketosis Ameliorates Renal Cyst Growth in Polycystic Kidney Disease". Cell Metabolism. 30 (6): 1007–1023.e5. doi:10.1016/j.cmet.2019.09.012. PMC 6904245. PMID 31631001.
- Reed B, Helal I, McFann K, Wang W, Yan XD, Schrier RW (July 2012). "The impact of type II diabetes mellitus in patients with autosomal dominant polycystic kidney disease". Nephrology, Dialysis, Transplantation. 27 (7): 2862–2865. doi:10.1093/ndt/gfr744. PMC 3398061. PMID 22207329.
- Nowak KL, You Z, Gitomer B, Brosnahan G, Torres VE, Chapman AB, et al. (February 2018). "Overweight and Obesity Are Predictors of Progression in Early Autosomal Dominant Polycystic Kidney Disease". Journal of the American Society of Nephrology. 29 (2): 571–578. doi:10.1681/ASN.2017070819. PMC 5791072. PMID 29118087.
- Kramers BJ, Koorevaar IW, Drenth JP, de Fijter JW, Neto AG, Peters DJ, et al. (October 2020). "Salt, but not protein intake, is associated with accelerated disease progression in autosomal dominant polycystic kidney disease". Kidney International. 98 (4): 989–998. doi:10.1016/j.kint.2020.04.053. hdl:2066/225147. PMID 32534051. S2CID 219637514.
- Kramers BJ, Koorevaar IW, Drenth JP, de Fijter JW, Neto AG, Peters DJ, et al. (October 2020). "Salt, but not protein intake, is associated with accelerated disease progression in autosomal dominant polycystic kidney disease". Kidney International. 98 (4): 989–998. doi:10.1016/j.kint.2020.04.053. hdl:2066/225147. PMID 32534051. S2CID 219637514.
- ^ Torres JA, Rezaei M, Broderick C, Lin L, Wang X, Hoppe B, et al. (July 2019). "Crystal deposition triggers tubule dilation that accelerates cystogenesis in polycystic kidney disease". The Journal of Clinical Investigation. 129 (10): 4506–4522. doi:10.1172/JCI128503. PMC 6763267. PMID 31361604.
- Amro OW, Paulus JK, Noubary F, Perrone RD (December 2016). "Low-Osmolar Diet and Adjusted Water Intake for Vasopressin Reduction in Autosomal Dominant Polycystic Kidney Disease: A Pilot Randomized Controlled Trial". American Journal of Kidney Diseases. 68 (6): 882–891. doi:10.1053/j.ajkd.2016.07.023. PMC 5123924. PMID 27663039.
- Allison SJ (December 2019). "Crystal deposition aids cystogenesis". Nature Reviews. Nephrology. 15 (12): 730. doi:10.1038/s41581-019-0215-7. PMID 31551591. S2CID 202733384.
- Tellman MW, Bahler CD, Shumate AM, Bacallao RL, Sundaram CP (May 2015). "Management of pain in autosomal dominant polycystic kidney disease and anatomy of renal innervation". The Journal of Urology. 193 (5): 1470–1478. doi:10.1016/j.juro.2014.10.124. hdl:1805/7798. PMID 25534330.
- ^ Mohsen T, Gomha MA (December 2005). "Treatment of symptomatic simple renal cysts by percutaneous aspiration and ethanol sclerotherapy". BJU International. 96 (9): 1369–1372. doi:10.1111/j.1464-410X.2005.05851.x. PMID 16287460.
- ^ Okeke AA, Mitchelmore AE, Keeley FX, Timoney AG (October 2003). "A comparison of aspiration and sclerotherapy with laparoscopic de-roofing in the management of symptomatic simple renal cysts". BJU International. 92 (6): 610–613. doi:10.1046/j.1464-410x.2003.04417.x. PMID 14511045.
- ^ Brown JA, Torres VE, King BF, Segura JW (July 1996). "Laparoscopic marsupialization of symptomatic polycystic kidney disease". The Journal of Urology. 156 (1): 22–27. doi:10.1016/s0022-5347(01)65927-5. PMID 8648810.
- ^ McDougall EM (December 2000). "Approach to decortication of simple cysts and polycystic kidneys". Journal of Endourology. 14 (10): 821–827. doi:10.1089/end.2000.14.821. PMID 11206615.
- Consonni P, Nava L, Scattoni V, Bianchi A, Spaliviero M, Guazzoni G, et al. (December 1996). "". Archivio Italiano di Urologia, Andrologia. 68 (5 Suppl): 27–30. PMID 9162369.
- Casteleijn NF, van Gastel MD, Blankestijn PJ, Drenth JP, de Jager RL, Leliveld AM, et al. (April 2017). "Novel treatment protocol for ameliorating refractory, chronic pain in patients with autosomal dominant polycystic kidney disease". Kidney International. 91 (4): 972–981. doi:10.1016/j.kint.2016.12.007. PMID 28159317.
- Nitschke AM, Ray CE (September 2013). "Percutaneous neurolytic celiac plexus block". Seminars in Interventional Radiology. 30 (3): 318–321. doi:10.1055/s-0033-1353485. PMC 3773031. PMID 24436554.
- Facciorusso, A.; Del Prete, V.; Antonino, M.; Buccino, V. R.; Muscatiello, N. (2019). "Response to repeat echoendoscopic celiac plexus neurolysis in pancreatic cancer patients: A machine learning approach". Pancreatology. 19 (6): 866–872. doi:10.1016/j.pan.2019.07.038. PMID 31375433. S2CID 199389236. Retrieved 5 January 2023.
- Alam A, Perrone RD (March 2010). "Management of ESRD in patients with autosomal dominant polycystic kidney disease". Advances in Chronic Kidney Disease. 17 (2): 164–172. doi:10.1053/j.ackd.2009.12.006. PMID 20219619.
- ^ Wagner MD, Prather JC, Barry JM (June 2007). "Selective, concurrent bilateral nephrectomies at renal transplantation for autosomal dominant polycystic kidney disease". The Journal of Urology. 177 (6): 2250–4, discussion 2254. doi:10.1016/j.juro.2007.01.146. PMID 17509331.
- ^ Cristea O, Yanko D, Felbel S, House A, Sener A, Luke PP (July 2014). "Maximal kidney length predicts need for native nephrectomy in ADPKD patients undergoing renal transplantation". Canadian Urological Association Journal. 8 (7–8): 278–282. doi:10.5489/cuaj.2128. PMC 4137014. PMID 25210553.
- Fuller TF, Brennan TV, Feng S, Kang SM, Stock PG, Freise CE (December 2005). "End stage polycystic kidney disease: indications and timing of native nephrectomy relative to kidney transplantation". The Journal of Urology. 174 (6): 2284–2288. doi:10.1097/01.ju.0000181208.06507.aa. PMID 16280813. S2CID 25363382.
- Cohen D, Timsit MO, Chrétien Y, Thiounn N, Vassiliu V, Mamzer MF, et al. (November 2008). "". Progres en Urologie. 18 (10): 642–649. doi:10.1016/j.purol.2008.06.004. PMID 18971106.
- Hajj P, Ferlicot S, Massoud W, Awad A, Hammoudi Y, Charpentier B, et al. (September 2009). "Prevalence of renal cell carcinoma in patients with autosomal dominant polycystic kidney disease and chronic renal failure". Urology. 74 (3): 631–634. doi:10.1016/j.urology.2009.02.078. PMID 19616833.
- ^ Courivaud C, Roubiou C, Delabrousse E, Bresson-Vautrin C, Chalopin JM, Ducloux D (April 2014). "Polycystic kidney size and outcomes on peritoneal dialysis: comparison with haemodialysis". Clinical Kidney Journal. 7 (2): 138–143. doi:10.1093/ckj/sft171. PMC 4377775. PMID 25852862.
- Nunes AC, Milani V, Porsch DB, Rossato LB, Mattos CB, Roisenberg I, Barros EJ (2008). "Frequency and clinical profile of patients with polycystic kidney disease in southern Brazil". Renal Failure. 30 (2): 169–173. doi:10.1080/08860220701810265. PMID 18300116.
- Bleyer AJ, Hart TC (June 2004). "Polycystic kidney disease". The New England Journal of Medicine. 350 (25): 2622, author reply 2622. doi:10.1056/NEJM200406173502519. PMID 15201424.
- Corradi V, Gastaldon F, Virzì GM, de Cal M, Soni S, Chionh C, et al. (October 2009). "Clinical pattern of adult polycystic kidney disease in a northeastern region of Italy". Clinical Nephrology. 72 (4): 259–267. doi:10.5414/CNP72259. PMID 19825331.
- Hamanoue S, Hoshino J, Suwabe T, Marui Y, Ueno T, Kikuchi K, et al. (June 2015). "Peritoneal Dialysis is Limited by Kidney and Liver Volume in Autosomal Dominant Polycystic Kidney Disease". Therapeutic Apheresis and Dialysis. 19 (3): 207–211. doi:10.1111/1744-9987.12272. PMID 25612237. S2CID 27836789.
- Matas AJ, Smith JM, Skeans MA, Lamb KE, Gustafson SK, Samana CJ, et al. (January 2013). "OPTN/SRTR 2011 Annual Data Report: kidney". American Journal of Transplantation. 13 (Suppl 1): 11–46. doi:10.1111/ajt.12019. PMC 5527691. PMID 23237695.
- ^ Cornec-Le Gall E, Le Meur Y (September 2014). "Polycystic kidney disease: Kidney volume--a crystal ball for ADPKD prognosis?". Nature Reviews. Nephrology. 10 (9): 485–486. doi:10.1038/nrneph.2014.132. PMID 25092148. S2CID 22042874.
- Kumar V, Abbas AK, Aster JC (2015). Robbins and Cotran Pathologic Basis of Disease (Ninth ed.). Philadelphia, PA. p. 947. ISBN 978-1-4557-2613-4. OCLC 879416939.
{{cite book}}: CS1 maint: location missing publisher (link)
External links
- "Polycystic Kidney Disease". National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). U.S. Department of Health and Human Services. Archived from the original on 2011-06-08.
- "Polycystic Kidney Disease". Genes and Disease . Bethesda (MD): National Center for Biotechnology Information (US). 1998.
| Classification | D |
|---|---|
| External resources |
| Congenital malformations and deformations of urinary system | |||||
|---|---|---|---|---|---|
| Abdominal |
| ||||
| Pelvic |
| ||||
| Both | |||||
| Vestigial |
| ||||
| Cystic diseases | |
|---|---|
| Respiratory system | |
| Skin |
|
| Human musculoskeletal system | |
| Human digestive system | |
| Nervous system | |
| Genitourinary system | |
| Other conditions | |