| Revision as of 14:43, 19 August 2022 edit2a02:85f:f50f:e78e:2d22:e06:cf2e:ee94 (talk) →Diagnosis← Previous edit | Revision as of 14:47, 19 August 2022 edit undo2a02:85f:f50f:e78e:2d22:e06:cf2e:ee94 (talk) →MicroRNA signatureNext edit → | ||
| Line 272: | Line 272: | ||
| === MicroRNA signature === | === MicroRNA signature === | ||
| A systematic review of miRNA levels in pediatric B-ALL compared with controls revealed that miR-155 is the most frequently reported upregulated miRNA (followed by miR-146a, miR-181b, miR-222 and miR-708; two citations for miR-16, miR-21, miR-34a, miR-100, miR-128-1, miR-181a, miR-181c, miR-195, miR-210, miR-320a and miR-660), whereas the most frequently reported downregulated miRNA is miR-374a (two citations for miR-27a, miR-30c, miR-196b, miR-223 and miR-494) <ref name=miRNAs_ALL>{{cite journal|vauthors= Kyriakidis I, Kyriakidis K, Tsezou A|title= MicroRNAs and the Diagnosis of Childhood Acute Lymphoblastic Leukemia: Systematic Review, Meta-Analysis and Re-Analysis with Novel Small RNA-Seq Tools | journal = Cancers | volume = 14 | issue = 16 | pages = 3976 | date = August 2022 | doi = 10.3390/cancers14163976 }}</ref>. | A systematic review of miRNA levels in pediatric B-ALL compared with controls revealed that miR-155 is the most frequently reported upregulated miRNA (followed by miR-146a, miR-181b, miR-222 and miR-708; two citations for miR-16, miR-21, miR-34a, miR-100, miR-128-1, miR-181a, miR-181c, miR-195, miR-210, miR-320a and miR-660), whereas the most frequently reported downregulated miRNA is miR-374a (two citations for miR-27a, miR-30c, miR-196b, miR-223 and miR-494). To date, there is no consensus miRNA signature and this is mainly attributed to different miRNA signatures across ALL subtypes and methodology of conducted studies <ref name=miRNAs_ALL>{{cite journal|vauthors= Kyriakidis I, Kyriakidis K, Tsezou A|title= MicroRNAs and the Diagnosis of Childhood Acute Lymphoblastic Leukemia: Systematic Review, Meta-Analysis and Re-Analysis with Novel Small RNA-Seq Tools | journal = Cancers | volume = 14 | issue = 16 | pages = 3976 | date = August 2022 | doi = 10.3390/cancers14163976 }}</ref>. | ||
| == Treatment == | == Treatment == | ||
Revision as of 14:47, 19 August 2022
Blood cancer characterised by overproduction of lymphoblastsMedical condition
| Acute lymphoblastic leukemia | |
|---|---|
| Other names | Acute lymphocytic leukemia, acute lymphoid leukemia |
 | |
| Bone marrow aspirate smear from a person with precursor B-cell ALL. The large purple cells are lymphoblasts. | |
| Specialty | Hematology, oncology |
| Symptoms | Feeling tired, pale color, fever, easy bleeding or bruising, bone pain, enlarged lymph nodes |
| Complications | Infection, tumor lysis syndrome |
| Usual onset | 2–5 years old |
| Types | B-cell ALL, T-cell ALL |
| Causes | Usually unknown |
| Risk factors | Identical twin with ALL, Down syndrome, Fanconi anemia, ataxia telangiectasia, Klinefelter syndrome, high birth weight, significant radiation exposure |
| Diagnostic method | Blood tests and bone marrow examination |
| Differential diagnosis | Infectious mononucleosis, acute myeloid leukemia, lymphoblastic lymphoma, aplastic anemia |
| Treatment | Chemotherapy, stem cell transplantation, radiation therapy, targeted therapy |
| Prognosis | Children: 90% five-year survival rate Adults: 35% five-year survival |
| Frequency | 1 in 1,750 children |
| Deaths | 111,000 (2015) |
Acute lymphoblastic leukemia (ALL) is a cancer of the lymphoid line of blood cells characterized by the development of large numbers of immature lymphocytes. Symptoms may include feeling tired, pale skin color, fever, easy bleeding or bruising, enlarged lymph nodes, or bone pain. As an acute leukemia, ALL progresses rapidly and is typically fatal within weeks or months if left untreated.
In most cases, the cause is unknown. Genetic risk factors may include Down syndrome, Li-Fraumeni syndrome, or neurofibromatosis type 1. Environmental risk factors may include significant radiation exposure or prior chemotherapy. Evidence regarding electromagnetic fields or pesticides is unclear. Some hypothesize that an abnormal immune response to a common infection may be a trigger. The underlying mechanism involves multiple genetic mutations that results in rapid cell division. The excessive immature lymphocytes in the bone marrow interfere with the production of new red blood cells, white blood cells, and platelets. Diagnosis is typically based on blood tests and bone marrow examination.
ALL is typically treated initially with chemotherapy aimed at bringing about remission. This is then followed by further chemotherapy typically over a number of years. Treatment usually also includes intrathecal chemotherapy since systemic chemotherapy can have limited penetration into the central nervous system and the central nervous system is a common site for relapse of acute lymphoblastic leukemia.
Treatment can also include radiation therapy if spread to the brain has occurred. Stem cell transplantation may be used if the disease recurs following standard treatment. Additional treatments such as Chimeric antigen receptor T cell immunotherapy are being used and further studied.
ALL affected about 876,000 people globally in 2015 and resulted in about 111,000 deaths. It occurs most commonly in children, particularly those between the ages of two and five. In the United States it is the most common cause of cancer and death from cancer among children. ALL is notable for being the first disseminated cancer to be cured. Survival for children increased from under 10% in the 1960s to 90% in 2015. Survival rates remain lower for babies (50%) and adults (35%). According to the National Cancer Intelligence Network (NCIN), generally for people with ALL: around 70 out of 100 people (70%) will survive their leukemia for 5 years or more after they are diagnosed.
Signs and symptoms
Initial symptoms can be nonspecific, particularly in children. Over 50% of children with leukemia had one or more of five features: a liver one can feel (64%), a spleen one can feel (61%), pale complexion (54%), fever (53%), and bruising (52%). Additionally, recurrent infections, feeling tired, arm or leg pain, and enlarged lymph nodes can be prominent features. The B symptoms, such as fever, night sweats, and weight loss, are often present as well.
Central nervous system (CNS) symptoms such as cranial neuropathies due to meningeal infiltration are identified in less than 10% of adults and less than 5% of children, particularly mature B-cell ALL (Burkitt leukemia) at presentation.
The signs and symptoms of ALL are variable and include:
- Generalized weakness and feeling tired
- Anemia
- Dizziness
- Headache, vomiting, lethargy, neck stiffness, or cranial nerve palsies (CNS involvement)
- Frequent or unexplained fever and infection
- Weight loss and/or loss of appetite
- Excessive and unexplained bruising
- Bone pain, joint pain (caused by the spread of "blast" cells to the surface of the bone or into the joint from the marrow cavity)
- Breathlessness
- Enlarged lymph nodes, liver, and/or spleen
- Pitting edema (swelling) in the lower limbs and/or abdomen
- Petechiae, which are tiny red spots or lines in the skin due to low platelet levels
- Testicular enlargement
- Mediastinal mass
Cause

The cancerous cell in ALL is the lymphoblast. Normal lymphoblasts develop into mature, infection-fighting B-cells or T-cells, also called lymphocytes. Signals in the body control the number of lymphocytes so neither too few nor too many are made. In ALL, both the normal development of some lymphocytes and the control over the number of lymphoid cells become defective.
ALL emerges when a single lymphoblast gains many mutations to genes that affect blood cell development and proliferation. In childhood ALL, this process begins at conception with the inheritance of some of these genes. These genes, in turn, increase the risk that more mutations will occur in developing lymphoid cells. Certain genetic syndromes, like Down Syndrome, have the same effect. Environmental risk factors are also needed to help create enough genetic mutations to cause disease. Evidence for the role of the environment is seen in childhood ALL among twins, where only 10–15% of both genetically identical twins get ALL. Since they have the same genes, different environmental exposures explain why one twin gets ALL and the other does not.
Infant ALL is a rare variant that occurs in babies less than one-year-old. KMT2A (formerly MLL) gene rearrangements are most common and occur in the embryo or fetus before birth. These rearrangements result in increased expression of blood cell development genes by promoting gene transcription and through epigenetic changes. In contrast to childhood ALL, environmental factors are not thought to play a significant role. Aside from the KMT2A rearrangement, only one extra mutation is typically found. Environmental exposures are not needed to help create more mutations.
Risk factors
Genetics
Common inherited risk factors include mutations in ARID5B, CDKN2A/2B, CEBPE, IKZF1, GATA3, PIP4K2A and, more rarely, TP53. These genes play important roles in cellular development, proliferation, and differentiation. Individually, most of these mutations are low risk for ALL. Significant risk of disease occurs when a person inherits several of these mutations together.
The uneven distribution of genetic risk factors may help explain differences in disease rates among ethnic groups. For instance, the ARID5B mutation is less common in ethnic African populations.
Several genetic syndrome also carry increased risk of ALL. These include: Down syndrome, Fanconi anemia, Bloom syndrome, X-linked agammaglobulinemia, severe combined immunodeficiency, Shwachman-Diamond syndrome, Kostmann syndrome, neurofibromatosis type 1, ataxia-telangiectasia, paroxysmal nocturnal hemoglobinuria, and Li-Fraumeni syndrome. Fewer than 5% of cases are associated with a known genetic syndrome.
Rare mutations in ETV6 and PAX5 are associated with a familial form of ALL with autosomal dominant patterns of inheritance.
Environmental
The environmental exposures that contribute to emergence of ALL is contentious and a subject of ongoing debate.
High levels of radiation exposure from nuclear fallout is a known risk factor for developing leukemia. Evidence whether lesser radiation, as from x-ray imaging during pregnancy, increases risk of disease remains inconclusive. Studies that have identified an association between x-ray imaging during pregnancy and ALL found only a slightly increased risk. Exposure to strong electromagnetic radiation from power lines has also been associated with a slightly increased risk of ALL. This result is questioned as no causal mechanism linking electromagnetic radiation with cancer is known.
High birth weight (greater than 4000g or 8.8lbs) is also associated with a small increased risk. The mechanism connecting high birth weight to ALL is also not known.
Evidence suggests that secondary leukemia can develop in individuals treated with certain types of chemotherapy, such as epipodophyllotoxins and cyclophosphamide.
Infections
There is some evidence that a common infection, such as influenza, may indirectly promote the emergence of ALL. The delayed-infection hypothesis states that ALL results from an abnormal immune response to infection in a person with genetic risk factors. Delayed development of the immune system due to limited disease exposure may result in excessive production of lymphocytes and increased mutation rate during an illness. Several studies have identified lower rates of ALL among children with greater exposure to illness early in life. Very young children who attend daycare have lower rates of ALL. Evidence from many other studies looking at disease exposure and ALL is inconclusive. Some researchers have linked the hygiene hypothesis.
Mechanism
Several characteristic genetic changes lead to the creation of a leukemic lymphoblast. These changes include chromosomal translocations, intrachromosomal rearrangements, changes in the number of chromosomes in leukemic cells, and additional mutations in individual genes. Chromosomal translocations involve moving a large region of DNA from one chromosome to another. This move can result in placing a gene from one chromosome that promotes cell division to a more actively transcribed area on another chromosome. The result is a cell that divides more often. An example of this includes the translocation of C-MYC, a gene that encodes a transcription factor that leads to increased cell division, next to the immunoglobulin heavy- or light-chain gene enhancers, leading to increased C-MYC expression and increased cell division. Other large changes in chromosomal structure can result in the placement of two genes directly next to each other. The result is the combination of two usually separate proteins into a new fusion protein. This protein can have a new function that promotes the development of cancer. Examples of this include the ETV6-RUNX1 fusion gene that combines two factors that promote blood cell development and the BCR-ABL1 fusion gene of the Philadelphia chromosome. BCR-ABL1 encodes an always-activated tyrosine kinase that causes frequent cell division. These mutations produce a cell that divides more often, even in the absence of growth factors.
Other genetic changes in B-cell ALL include changes to the number of chromosomes within the leukemic cells. Gaining at least five additional chromosomes, called high hyperdiploidy, occurs more commonly. Less often, chromosomes are lost, called hypodiploidy, which is associated with a poorer prognosis. Additional common genetic changes in B-cell ALL involve non-inherited mutations to PAX5 and IKZF1. In T-cell ALL, LYL1, TAL1, TLX1, and TLX3 rearrangements can occur.
ALL results when enough of these genetic changes are present in a single lymphoblast. In childhood ALL, for example, one fusion gene translocation is often found along with six to eight other ALL-related genetic changes. The initial leukemic lymphoblast copies itself into an excessive number of new lymphoblasts, none of which can develop into functioning lymphocytes. These lymphoblasts build up in the bone marrow and may spread to other sites in the body, such as lymph nodes, the mediastinum, the spleen, the testicles, and the brain, leading to the common symptoms of the disease.
Diagnosis
Diagnosing ALL begins with a thorough medical history, physical examination, complete blood count, and blood smears. While many symptoms of ALL can be found in common illnesses, persistent or unexplained symptoms raise suspicion of cancer. Because many features on the medical history and exam are not specific to ALL, further testing is often needed. A large number of white blood cells and lymphoblasts in the circulating blood can be suspicious for ALL because they indicate a rapid production of lymphoid cells in the marrow. The higher these numbers typically point to a worse prognosis. While white blood cell counts at initial presentation can vary significantly, circulating lymphoblast cells are seen on peripheral blood smears in the majority of cases.
A bone marrow biopsy provides conclusive proof of ALL, typically with >20% of all cells being leukemic lymphoblasts. A lumbar puncture (also known as a spinal tap) can determine whether the spinal column and brain have been invaded. Brain and spinal column involvement can be diagnosed either through confirmation of leukemic cells in the lumbar puncture or through clinical signs of CNS leukemia as described above. Laboratory tests that might show abnormalities include blood count, kidney function, electrolyte, and liver enzyme tests.
Pathological examination, cytogenetics (in particular the presence of Philadelphia chromosome), and immunophenotyping establish whether the leukemic cells are myeloblastic (neutrophils, eosinophils, or basophils) or lymphoblastic (B lymphocytes or T lymphocytes). Cytogenetic testing on the marrow samples can help classify disease and predict how aggressive the disease course will be. Different mutations have been associated with shorter or longer survival. Immunohistochemical testing may reveal TdT or CALLA antigens on the surface of leukemic cells. TdT is a protein expressed early in the development of pre-T and pre-B cells, whereas CALLA is an antigen found in 80% of ALL cases and also in the "blast crisis" of CML.
Medical imaging (such as ultrasound or CT scanning) can find invasion of other organs commonly the lung, liver, spleen, lymph nodes, brain, kidneys, and reproductive organs.
-
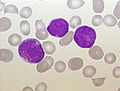 acute lymphoblastic leukemia (ALL), peripheral blood of a child, Pappenheim stain, magnification x100
acute lymphoblastic leukemia (ALL), peripheral blood of a child, Pappenheim stain, magnification x100
-
 bone marrow smear (large magnification) from a person with acute lymphoblastic leukemia
bone marrow smear (large magnification) from a person with acute lymphoblastic leukemia
-
 bone marrow smear from a person with acute lymphoblastic leukemia
bone marrow smear from a person with acute lymphoblastic leukemia



Immunophenotyping
In addition to cell morphology and cytogenetics, immunophenotyping, a laboratory technique used to identify proteins that are expressed on their cell surface, is a key component in the diagnosis of ALL. The preferred method of immunophenotyping is through flow cytometry. In the malignant lymphoblasts of ALL, expression of terminal deoxynucleotidyl transferase (TdT) on the cell surface can help differentiate malignant lymphocyte cells from reactive lymphocytes, white blood cells that are reacting normally to an infection in the body. On the other hand, myeloperoxidase (MPO), a marker for the myeloid lineage, is typically not expressed. Because precursor B cell and precursor T cells look the same, immunophenotyping can help differentiate the subtype of ALL and the level of maturity of the malignant white blood cells. The subtypes of ALL as determined by immunophenotype and according to the stages of maturation.
| B cell Lineage | T cell Lineage |
|---|---|
| pre-pre-B ALL (pro-B-ALL) | precursor T- ALL |
| common ALL | mature T-cell ALL |
| pre-B ALL | |
| mature B-cell ALL (Burkitt leukemia – FAB L3) |
An extensive panel of monoclonal antibodies to cell surface markers, particularly CD or cluster of differentiation markers, are used to classify cells by lineage. Below are immunological markers associated with B cell and T cell ALL.
| Immunological Markers | B cell Lineage | T cell Lineage |
|---|---|---|
| B cell Lineage | ||
| CD19, CD22, CD79a | + | − |
| CD10 | − or + (common ALL) | |
| cytoplasmic Ig | − or + (pre-B ALL) | |
| surface Ig | − or + (mature B-cell ALL) | |
| TdT | + | + |
| T cell Lineage | ||
| CD2, CD3, CD4, CD5, CD7, CD8 | − | + |
| TdT | + | + |
Cytogenetics
Cytogenetic analysis has shown different proportions and frequencies of genetic abnormalities in cases of ALL from different age groups. This information is particularly valuable for classification and can in part explain the different prognoses of these groups. In regards to genetic analysis, cases can be stratified according to ploidy, a number of sets of chromosomes in the cell, and specific genetic abnormalities, such as translocations. Hyperdiploid cells are defined as cells with more than 50 chromosomes, while hypodiploid are defined as cells with less than 44 chromosomes. Hyperdiploid cases tend to carry a good prognosis while hypodiploid cases do not. For example, the most common specific abnormality in childhood B-ALL is the t(12;21) ETV6-RUNX1 translocation, in which the RUNX1 gene, encoding a protein involved in transcriptional control of hemopoiesis, has been translocated and repressed by the ETV6-RUNX1 fusion protein.
Below is a table with the frequencies of some cytogenetic translocations and molecular genetic abnormalities in ALL.
| Cytogenetic translocation | Molecular genetic abnormality | % |
|---|---|---|
| cryptic t(12;21) | TEL–AML1 fusion | 25.4% |
| t(1;19)(q23;p13) | E2A–PBX (PBX1) fusion | 4.8% |
| t(9;22)(q34;q11) | BCR-ABL fusion(P185) | 1.6% |
| t(4;11)(q21;q23) | MLL–AF4 fusion | 1.6% |
| t(8;14)(q24;q32) | IGH-MYC fusion | |
| t(11;14)(p13;q11) | TCR–RBTN2 fusion |
Classification
French-American-British
Historically, prior to 2008, ALL was classified morphologically using the French-American-British (FAB) system that heavily relied on morphological assessment. The FAB system takes into account information on size, cytoplasm, nucleoli, basophilia (color of cytoplasm), and vacuolation (bubble-like properties).
| FAB Subtype | Cell Type | Characteristics | Comments |
|---|---|---|---|
| ALL - L1 | T cell or pre-B cell | Small and homogeneous (uniform) cells | |
| ALL - L2 | T cell or pre-B cell | Large and heterogeneous (varied) cells | |
| ALL - L3 | B cell | Large and varied cells with vacuoles | Mature B-cell ALL also named Burkitt leukemia. Typically, poor prognosis with standard therapy |
While some clinicians still use the FAB scheme to describe tumor cell appearance, much of this classification has been abandoned because of its limited impact on treatment choice and prognostic value.
World Health Organization
In 2008, the World Health Organization classification of acute lymphoblastic leukemia was developed in an attempt to create a classification system that was more clinically relevant and could produce meaningful prognostic and treatment decisions. This system recognized differences in genetic, immunophenotype, molecular, and morphological features found through cytogenetic and molecular diagnostics tests. This subtyping helps determine the prognosis and the most appropriate treatment for each specific case of ALL.
The WHO subtypes related to ALL are:
- B-lymphoblastic leukemia/lymphoma
- Not otherwise specified (NOS)
- with recurrent genetic abnormalities
- with t(9;22)(q34.1;q11.2);BCR-ABL1
- with t(v;11q23.3);KMT2A rearranged
- with t(12;21)(p13.2;q22.1); ETV6-RUNX1
- with t(5;14)(q31.1;q32.3) IL3-IGH
- with t(1;19)(q23;p13.3);TCF3-PBX1
- with hyperdiploidy
- with hypodiploidy
- T-lymphoblastic leukemia/lymphoma
- Acute leukemias of ambiguous lineage
- Acute undifferentiated leukemia
- Mixed phenotype acute leukemia (MPAL) with t(9;22)(q34.1;q11.2); BCR-ABL1
- MPAL with t(v;11q23.3); KMT2A rearranged
- MPAL, B/myeloid, NOS
- MPAL, T/myeloid, NOS
MicroRNA signature
A systematic review of miRNA levels in pediatric B-ALL compared with controls revealed that miR-155 is the most frequently reported upregulated miRNA (followed by miR-146a, miR-181b, miR-222 and miR-708; two citations for miR-16, miR-21, miR-34a, miR-100, miR-128-1, miR-181a, miR-181c, miR-195, miR-210, miR-320a and miR-660), whereas the most frequently reported downregulated miRNA is miR-374a (two citations for miR-27a, miR-30c, miR-196b, miR-223 and miR-494). To date, there is no consensus miRNA signature and this is mainly attributed to different miRNA signatures across ALL subtypes and methodology of conducted studies .
Treatment

The aim of treatment is to induce a lasting remission, defined as the absence of detectable cancer cells in the body (usually less than 5% blast cells in the bone marrow).
Over the past several decades, there have been strides to increase the efficacy of treatment regimens, resulting in increased survival rates. Possible treatments for acute leukemia include chemotherapy, steroids, radiation therapy, intensive combined treatments (including bone marrow or stem cell transplants), targeted therapy, and/or growth factors.
Chemotherapy
Chemotherapy is the initial treatment of choice, and most people with ALL receive a combination of medications. There are no surgical options because of the body-wide distribution of the malignant cells. In general, cytotoxic chemotherapy for ALL combines multiple antileukemic drugs tailored to each person. Chemotherapy for ALL consists of three phases: remission induction, intensification, and maintenance therapy.
| Phase | Description | Agents |
|---|---|---|
| Remission induction | Aim to:
Must monitor closely for tumor lysis syndrome after initiating therapy Monitoring initial response to treatment is important as failure to show clearance of blood or bone marrow blasts within the first 2 weeks of therapy has been associated with a higher risk of relapse
Start CNS prophylaxis and administer intrathecal chemotherapy via Ommaya reservoir or multiple lumbar punctures |
Combination of:
Central nervous system prophylaxis can be achieved via:
In Philadelphia chromosome-positive ALL, the intensity of initial induction treatment may be less than has been traditionally given. |
| Consolidation/intensification | Use high doses of chemotherapy to further reduce tumor burden | Typical protocols use the following given as blocks (varies from 1-3 blocks depending on person's risk category) in different multi-drug combinations:
Central nervous system relapse is treated with intrathecal administration of hydrocortisone, methotrexate, and cytarabine. |
| Maintenance therapy | Kill any residual cell that was not killed by remission induction and intensification regimens
|
Typical protocol would include:
|
Due to the presence of CNS involvement in 10–40% of adults with ALL at diagnosis, most providers start Central nervous system (CNS) prophylaxis and treatment during the induction phase, and continue it during the consolidation/intensification period.
Adult chemotherapy regimens mimic those of childhood ALL; however, are linked with a higher risk of disease relapse with chemotherapy alone. It should be known that 2 subtypes of ALL (B-cell ALL and T-cell ALL) require special considerations when it comes to selecting an appropriate treatment regimen in adults with ALL. B-cell ALL is often associated with cytogenetic abnormalities (specifically, t(8;14), t (2;8), and t(8;22)), which require aggressive therapy consisting of brief, high-intensity regimens. T-cell ALL responds to cyclophosphamide-containing agents the most.
As the chemotherapy regimens can be intensive and protracted, many people have an intravenous catheter inserted into a large vein (termed a central venous catheter or a Hickman line), or a Portacath, usually placed near the collar bone, for lower infection risks and the long-term viability of the device. Males usually endure a longer course of treatment than females as the testicles can act as a reservoir for cancer.
Radiation therapy
Radiation therapy (or radiotherapy) is used on painful bony areas, in high disease burdens, or as part of the preparations for a bone marrow transplant (total body irradiation). In the past, physicians commonly utilized radiation in the form of whole-brain radiation for central nervous system prophylaxis, to prevent the occurrence and/or recurrence of leukemia in the brain. Recent studies showed that CNS chemotherapy provided results as favorable but with fewer developmental side effects. As a result, the use of whole-brain radiation has been more limited. Most specialists in adult leukemia have abandoned the use of radiation therapy for CNS prophylaxis, instead using intrathecal chemotherapy.
Biological therapy
Selection of biological targets on the basis of their combinatorial effects on the leukemic lymphoblasts can lead to clinical trials for improvement in the effects of ALL treatment. Tyrosine-kinase inhibitors (TKIs), such as imatinib, are often incorporated into the treatment plan for people with Bcr-Abl1+ (Ph+) ALL. However, this subtype of ALL is frequently resistant to the combination of chemotherapy and TKIs and allogeneic stem cell transplantation is often recommended upon relapse.
Immunotherapy
Chimeric antigen receptors (CARs) have been developed as a promising immunotherapy for ALL. This technology uses a single chain variable fragment (scFv) designed to recognize the cell surface marker CD19 as a method of treating ALL.
CD19 is a molecule found on all B-cells and can be used as a means of distinguishing the potentially malignant B-cell population. In this therapy, mice are immunized with the CD19 antigen and produce anti-CD19 antibodies. Hybridomas developed from mouse spleen cells fused to a myeloma cell line can be developed as a source for the cDNA encoding the CD19 specific antibody. The cDNA is sequenced and the sequence encoding the variable heavy and variable light chains of these antibodies are cloned together using a small peptide linker. This resulting sequence encodes the scFv. This can be cloned into a transgene, encoding what will become the endodomain of the CAR. Varying arrangements of subunits serve as the endodomain, but they generally consist of the hinge region that attaches to the scFv, a transmembrane region, the intracellular region of a costimulatory molecule such as CD28, and the intracellular domain of CD3-zeta containing ITAM repeats. Other sequences frequently included are: 4-1bb and OX40. The final transgene sequence, containing the scFv and endodomain sequences is then inserted into immune effector cells that are obtained from the person and expanded in vitro. In trials these have been a type of T-cell capable of cytotoxicity.
Inserting the DNA into the effector cell can be accomplished by several methods. Most commonly, this is done using a lentivirus that encodes the transgene. Pseudotyped, self-inactivating lentiviruses are an effective method for the stable insertion of a desired transgene into the target cell. Other methods include electroporation and transfection, but these are limited in their efficacy as transgene expression diminishes over time.
The gene-modified effector cells are then transplanted back into the person. Typically this process is done in conjunction with a conditioning regimen such as cyclophosphamide, which has been shown to potentiate the effects of infused T-cells. This effect has been attributed to making an immunologic space within which the cells populate. The process as a whole result in an effector cell, typically a T-cell, that can recognize a tumor cell antigen in a manner that is independent of the major histocompatibility complex and which can initiate a cytotoxic response.
In 2017, tisagenlecleucel was approved by the FDA as a CAR-T therapy for people with acute B-cell lymphoblastic leukaemia who did not respond adequately to other treatments or have relapsed. In a 22-day process, the "drug" is customized for each person. T cells purified from each person are modified by a virus that inserts genes that encode a chimaeric antigen receptor into their DNA, one that recognizes leukemia cells.
Relapsed ALL
Typically, people who experience a relapse in their ALL after initial treatment have a poorer prognosis than those who remain in complete remission after induction therapy. It is unlikely that recurrent leukemia will respond favorably to the standard chemotherapy regimen that was initially implemented, and instead, these people should be trialed on reinduction chemotherapy followed by allogeneic bone marrow transplantation. These people in relapse may also receive blinatumomab, as it has shown to increase remission rates and overall survival rates, without increased toxic effects.
Low dose palliative radiation may also help reduce the burden of tumor inside or outside the central nervous system and alleviate some symptoms.
Recently, there has also been evidence and approval of use for dasatinib, a tyrosine kinase inhibitor. It has shown efficacy in cases of people with Ph1-positive and imatinib-resistant ALL, but more research needs to be done on long-term survival and time to relapse.
Side effects
Chemotherapies or stem cell transplantations may require a platelet transfusion to prevent bleeding. Moreover, patients undergoing a stem cell transplantation can develop a graft-versus-host disease (GvHD). It was evaluated whether mesenchymal stromal cells can be used to prevent a GvHD. The evidence is very uncertain about the therapeutic effect of mesenchymal stromal cells to treat graft-versus-host diseases after a stem cell transplantation on the all-cause mortality and complete disappear of chronic acute graft-versus-host diseases. Mesenchymal stromal cells may results in little to no difference in the all-cause mortality, relapse of malignant disease and incidence of acute and chronic graft-versus-host diseases if they are used for prophylactic reason.
Supportive therapy
Adding physical exercises to the standard treatment for adult patients with haematological malignancies like ALL may result in little to no difference in mortality, the quality of life, and physical functioning. These exercises may result in a slight reduction in depression. Furthermore, aerobic physical exercises probably reduce fatigue. The evidence is very uncertain about the effect on anxiety and serious adverse events.
Gene therapy
Brexucabtagene autoleucel (Tecartus) was approved for the treatment of adults with relapsed or refractory B-cell precursor acute lymphoblastic leukemia in October 2021.
Each dose of brexucabtagene autoleucel is a customized treatment created using the recipient's own immune system to help fight the lymphoma. The recipient's T cells, a type of white blood cell, are collected and genetically modified to include a new gene that facilitates the targeting and killing of the lymphoma cells. These modified T cells are then infused back into the recipient.
Prognosis
Prior to the development of chemotherapy regimens and hematopoietic stem cell transplant, children were surviving a median length of 3 months, largely due to either infection or bleeding. Since the advent of chemotherapy, the prognosis for childhood leukemia has improved greatly and children with ALL are estimated to have a 95% probability of achieving a successful remission after 4 weeks of initiating treatment. People in pediatric care with ALL in developed countries have a greater than 80% five-year survival rate. It is estimated that 60–80% of adults undergoing induction chemotherapy achieve complete remission after 4 weeks, and those over the age of 70 have a cure rate of 5%.

However, there are differing prognoses for ALL among individuals depending on a variety of factors:
- Gender: Females tend to fare better than males.
- Ethnicity: Caucasians are more likely to develop acute leukemia than African-Americans, Asians, or Hispanics. However, they also tend to have a better prognosis than non-Caucasians.
- Age at diagnosis: children 1–10 years of age are most likely to develop ALL and to be cured of it. Cases in older people are more likely to result from chromosomal abnormalities (e.g., the Philadelphia chromosome) that make treatment more difficult and prognoses poorer. Older people are also likely to have co-morbid medical conditions that make it even more difficult to tolerate ALL treatment.
- White blood cell count at diagnosis of greater than 30,000 (B-ALL) or 100,000 (T-ALL) is associated with worse outcomes
- Cancer spreading into the Central nervous system (brain or spinal cord) has worse outcomes.
- Morphological, immunological, and genetic subtypes
- Person's response to initial treatment and longer length of time required (greater than 4 weeks) to reach complete remission
- Early relapse of ALL
- Minimal residual disease
- Genetic disorders, such as Down syndrome, and other chromosomal abnormalities (aneuoploidy and translocations)
| Factor | Unfavorable | Favorable |
|---|---|---|
| Age | <2 or >10 years | 3–5 years |
| Sex | Male | Female |
| Race | Black | Caucasian |
| Organomegaly | Present | Absent |
| Mediastinal mass | Present | Absent |
| CVS involvement | Present | Absent |
| Leukocyte count | B-ALL >30,000mm T-ALL >100,000mm | Low |
| Hemogblobin concentration | >10g/dl | <10g/dl |
| Cell type | Non Lymphoid | Lymphoid |
| Cell lineage | Pre B cell +
T-ALL (children) |
Early Pre B cell |
| Karyotype | Translocation | Hyperdiploidy |
| Response to treatment | Slow
>1 week to clear blasts from blood |
Rapid
<1 week to clear blasts from blood |
| Time to remission | >4 weeks | <4 weeks |
| Minimal residual disease | Positive at 3 – 6 months | Negative at 1 month (children) or 3 months (adults) |
Cytogenetics, the study of characteristic large changes in the chromosomes of cancer cells, is an important predictor of outcome. Some cytogenetic subtypes have a worse prognosis than others. These include:
- Person with t(9,22) positive-ALL (30% of adult ALL cases) and other Bcr-abl-rearranged leukemias are more likely to have a poor prognosis, but survival rates may rise with treatment consisting of chemotherapy and Bcr-abl tyrosine kinase inhibitors.
- A translocation between chromosomes 4 and 11 occurs in about 4% of cases and is most common in infants under 12 months.
| Cytogenetic change | Risk category |
|---|---|
| Philadelphia chromosome | Poor prognosis |
| t(4;11)(q21;q23) | Poor prognosis |
| t(8;14)(q24.1;q32) | Poor prognosis |
| Complex karyotype (more than four abnormalities) | Poor prognosis |
| Low hypodiploidy or near triploidy | Poor prognosis |
| Deletion of chromosome 7 | Poor prognosis |
| Trisomy 8 | Poor prognosis |
| High hyperdiploidy (trisomy 4, 10, 17) | Good prognosis |
| del(9p) | Good prognosis |
- Hyperdiploidy (>50 chromosomes) and t(12;21) are good prognostic factors and also makeup 50% of pediatric ALL cases.
| Prognosis | Cytogenetic findings |
|---|---|
| Favorable | Hyperdiploidy > 50 ; t (12;21) |
| Intermediate | Hyperdiploidy 47–50; Normal(diploidy); del (6q); Rearrangements of 8q24 |
| Unfavorable | Hypodiploidy-near haploidy; Near tetraploidy; del (17p); t (9;22); t (11q23) |
Unclassified ALL is considered to have an intermediate prognosis risk, somewhere in-between the good and poor risk categories.
Epidemiology
ALL affected about 876,000 people and resulted in 111,000 deaths globally in 2015. It occurs in both children and adults with highest rates seen between the ages three and seven years. Around 75% of cases occur before the age of 6 with a secondary rise after the age of 40. It is estimated to affect 1 in 1500 children.
Accounting for the broad age profiles of those affected, ALL newly occurs in about 1.7 per 100,000 people per year. ALL represents approximately 20% of adults and 80% of childhood leukemias, making it the most common childhood cancer. Although 80 to 90% of children will have a long term complete response with treatment, it remains the leading cause of cancer-related deaths among children. 85% of cases are of B-cell lineage and have an equal number of cases in both males and females. The remaining 15% of T-cell lineage have a male predominance.
Globally, ALL typically occurs more often in Caucasians, Hispanics, and Latin Americans than in Africans. In the US, ALL is more common in children from Caucasian (36 cases/million) and Hispanic (41 cases/million) descent when compared to those from African (15 cases/million) descent.
Pregnancy
Leukemia is rarely associated with pregnancy, affecting only about 1 in 10,000 pregnant women. The management of leukemia in a pregnant woman depends primarily on the type of leukemia. Acute leukemias normally require prompt, aggressive treatment, despite significant risks of pregnancy loss and birth defects, especially if chemotherapy is given during the developmentally sensitive first trimester.
References
- ^ "Childhood Acute Lymphoblastic Leukemia Treatment". National Cancer Institute. 8 December 2017. Retrieved 20 December 2017.
- ^ Hunger SP, Mullighan CG (October 2015). "Acute Lymphoblastic Leukemia in Children". The New England Journal of Medicine. 373 (16): 1541–52. doi:10.1056/nejmra1400972. PMID 26465987. S2CID 609394.
- ^ Ferri, Fred F. (2017). Ferri's Clinical Advisor 2018 E-Book: 5 Books in 1. Elsevier Health Sciences. p. 743. ISBN 9780323529570.
- ^ Inaba H, Greaves M, Mullighan CG (June 2013). "Acute lymphoblastic leukaemia". Lancet. 381 (9881): 1943–55. doi:10.1016/S0140-6736(12)62187-4. PMC 3816716. PMID 23523389.
- ^ Baljevic M, Jabbour E, O'Brien S, Kantarjian HM (2016). "Acute Lymphoblastic Leukemia". In Kantarjian HM, Wolff RA (eds.). The MD Anderson Manual of Medical Oncology (3 ed.). New York: McGraw-Hill Education. Retrieved 22 November 2017.
- ^ Childhood acute lymphoblastic leukemia. Vora, Ajay (editor). Cham, Switzerland: Springer International Publishing. 2017. pp. 1–44, 61–86. ISBN 9783319397078. OCLC 984342596.
{{cite book}}: CS1 maint: others (link) - ^ Cordo V, Meijerink J (January 2021). "T-cell Acute Lymphoblastic Leukemia: A Roadmap to Targeted Therapies". Blood Cancer Discovery. 2 (1): 19–31. doi:10.1158/2643-3230.BCD-20-0093. PMC 8447273. PMID 34661151.
- ^ Paul S, Kantarjian H, Jabbour EJ (November 2016). "Adult Acute Lymphoblastic Leukemia". Mayo Clinic Proceedings. 91 (11): 1645–1666. doi:10.1016/j.mayocp.2016.09.010. PMID 27814839.
- ^ Boer JM, den Boer ML (September 2017). "BCR-ABL1-like acute lymphoblastic leukaemia: From bench to bedside". European Journal of Cancer. 82: 203–218. doi:10.1016/j.ejca.2017.06.012. PMID 28709134.
- ^ Wang, Haidong; Naghavi, Mohsen; Allen, Christine; Barber, Ryan M.; Bhutta, Zulfiqar A.; Carter, Austin; Casey, Daniel C.; Charlson, Fiona J.; Chen, Alan Zian; Coates, Matthew M.; Coggeshall, Megan; Dandona, Lalit; Dicker, Daniel J.; Erskine, Holly E.; Ferrari, Alize J.; Fitzmaurice, Christina; Foreman, Kyle; Forouzanfar, Mohammad H.; Fraser, Maya S.; Fullman, Nancy; Gething, Peter W.; Goldberg, Ellen M.; Graetz, Nicholas; Haagsma, Juanita A.; Hay, Simon I.; Huynh, Chantal; Johnson, Catherine O.; Kassebaum, Nicholas J.; Kinfu, Yohannes; et al. (October 2016). "Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980-2015: a systematic analysis for the Global Burden of Disease Study 2015". Lancet. 388 (10053): 1459–1544. doi:10.1016/s0140-6736(16)31012-1. PMC 5388903. PMID 27733281.
- Marino BS, Fine KS (2013). Blueprints Pediatrics. Lippincott Williams & Wilkins. p. 205. ISBN 9781451116045.
- Pui, Ching-Hon; Evans, William E. (12 January 2006). "Treatment of Acute Lymphoblastic Leukemia". New England Journal of Medicine. 354 (2): 166–178. doi:10.1056/NEJMra052603. ISSN 0028-4793. PMID 16407512.
- Larson, Richard A. (2 January 2018). "Managing CNS disease in adults with acute lymphoblastic leukemia". Leukemia & Lymphoma. 59 (1): 3–13. doi:10.1080/10428194.2017.1326597. ISSN 1042-8194. PMID 28535095. S2CID 24564241.
- ^ Vos, Theo; Allen, Christine; Arora, Megha; Barber, Ryan M.; Bhutta, Zulfiqar A.; Brown, Alexandria; Carter, Austin; Casey, Daniel C.; Charlson, Fiona J.; Chen, Alan Z.; Coggeshall, Megan; Cornaby, Leslie; Dandona, Lalit; Dicker, Daniel J.; Dilegge, Tina; Erskine, Holly E.; Ferrari, Alize J.; Fitzmaurice, Christina; Fleming, Tom; Forouzanfar, Mohammad H.; Fullman, Nancy; Gething, Peter W.; Goldberg, Ellen M.; Graetz, Nicholas; Haagsma, Juanita A.; Hay, Simon I.; Johnson, Catherine O.; Kassebaum, Nicholas J.; Kawashima, Toana; et al. (October 2016). "Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015". Lancet. 388 (10053): 1545–1602. doi:10.1016/S0140-6736(16)31678-6. PMC 5055577. PMID 27733282.
- "Acute Lymphocytic Leukemia - Cancer Stat Facts". SEER. Retrieved 20 December 2017.
- ^ Tubergen DG, Bleyer A, Ritchey AK (2011). "Acute Lymphoblastic Leukemia". In Kliegman RM, Stanton BM, Geme J, Schor NF, Behrman RE (eds.). Nelson Textbook of Pediatrics (19th ed.). Philadelphia, PA: Elsevier/Saunders. pp. 1732–1737. ISBN 978-1437707557. OCLC 706780860.
- Brown P (6 December 2013). "Treatment of infant leukemias: challenge and promise". Hematology. American Society of Hematology. Education Program. 2013 (1): 596–600. doi:10.1182/asheducation-2013.1.596. PMC 4729208. PMID 24319237.
- Clarke RT, Van den Bruel A, Bankhead C, Mitchell CD, Phillips B, Thompson MJ (October 2016). "Clinical presentation of childhood leukaemia: a systematic review and meta-analysis". Archives of Disease in Childhood. 101 (10): 894–901. doi:10.1136/archdischild-2016-311251. PMID 27647842.
- "Acute Lymphoblastic Leukemia". The Lecturio Medical Concept Library. Retrieved 27 June 2021.
- Cortes J (February 2001). "Central nervous system involvement in adult acute lymphocytic leukemia". Hematology/Oncology Clinics of North America. 15 (1): 145–62. doi:10.1016/s0889-8588(05)70203-3. PMID 11253605.
- ^ Acute Lymphoblastic Leukemia at eMedicine
- Bleyer WA (August 1988). "Central nervous system leukemia". Pediatric Clinics of North America. 35 (4): 789–814. doi:10.1016/s0031-3955(16)36510-5. PMID 3047654.
- Ingram LC, Fairclough DL, Furman WL, Sandlund JT, Kun LE, Rivera GK, Pui CH (May 1991). "Cranial nerve palsy in childhood acute lymphoblastic leukemia and non-Hodgkin's lymphoma". Cancer. 67 (9): 2262–8. doi:10.1002/1097-0142(19910501)67:9<2262::aid-cncr2820670909>3.0.co;2-u. PMID 2013032.
- Terwilliger T, Abdul-Hay M (June 2017). "Acute lymphoblastic leukemia: a comprehensive review and 2017 update". Blood Cancer Journal. 7 (6): e577. doi:10.1038/bcj.2017.53. PMC 5520400. PMID 28665419.
- Meyer C, Hofmann J, Burmeister T, Gröger D, Park TS, Emerenciano M, et al. (November 2013). "The MLL recombinome of acute leukemias in 2013". Leukemia. 27 (11): 2165–76. doi:10.1038/leu.2013.135. PMC 3826032. PMID 23628958.
- Benedikt A, Baltruschat S, Scholz B, Bursen A, Arrey TN, Meyer B, et al. (January 2011). "The leukemogenic AF4-MLL fusion protein causes P-TEFb kinase activation and altered epigenetic signatures". Leukemia. 25 (1): 135–44. doi:10.1038/leu.2010.249. PMID 21030982.
- Preston DL, Kusumi S, Tomonaga M, Izumi S, Ron E, Kuramoto A, et al. (February 1994). "Cancer incidence in atomic bomb survivors. Part III. Leukemia, lymphoma and multiple myeloma, 1950-1987". Radiation Research. 137 (2 Suppl): S68-97. Bibcode:1994RadR..137S..68P. doi:10.2307/3578893. JSTOR 3578893. PMID 8127953.
- Smith MA, Rubinstein L, Anderson JR, Arthur D, Catalano PJ, Freidlin B, et al. (February 1999). "Secondary leukemia or myelodysplastic syndrome after treatment with epipodophyllotoxins". Journal of Clinical Oncology. 17 (2): 569–77. doi:10.1200/JCO.1999.17.2.569. PMID 10080601.
- Greaves M (August 2018). "A causal mechanism for childhood acute lymphoblastic leukaemia". Nature Reviews. Cancer. 18 (8): 471–484. doi:10.1038/s41568-018-0015-6. PMC 6986894. PMID 29784935.
- Collier, J.A.B (1991). Oxford Handbook of Clinical Specialties, Third Edition. Oxford. p. 810. ISBN 978-0-19-262116-0.
- Longo, D (2011). "Chapter 110: Malignancies of Lymphoid Cells". Harrison's Principles of Internal Medicine (18 ed.). New York: McGraw-Hill Professional. ISBN 978-0-07-174889-6.
- Rytting, ME, ed. (November 2013). "Acute Leukemia". Merck Manual Professional. Merck Sharp & Dohme Corp. Archived from the original on 15 July 2014. Retrieved 17 April 2014.
- ^ Hoffbrand AV, Moss PA (6 October 2015). Hoffbrand's essential haematology (Seventh ed.). Chichester, West Sussex. ISBN 9781118408636. OCLC 910009732.
{{cite book}}: CS1 maint: location missing publisher (link) - Bhojwani D, Pei D, Sandlund JT, Jeha S, Ribeiro RC, Rubnitz JE, et al. (February 2012). "ETV6-RUNX1-positive childhood acute lymphoblastic leukemia: improved outcome with contemporary therapy". Leukemia. 26 (2): 265–70. doi:10.1038/leu.2011.227. PMC 3345278. PMID 21869842.
- Stams WA, den Boer ML, Beverloo HB, Meijerink JP, van Wering ER, Janka-Schaub GE, Pieters R (April 2005). "Expression levels of TEL, AML1, and the fusion products TEL-AML1 and AML1-TEL versus drug sensitivity and clinical outcome in t(12;21)-positive pediatric acute lymphoblastic leukemia". Clinical Cancer Research. 11 (8): 2974–80. doi:10.1158/1078-0432.CCR-04-1829. PMID 15837750.
- ^ Pakakasama S, Kajanachumpol S, Kanjanapongkul S, Sirachainan N, Meekaewkunchorn A, Ningsanond V, Hongeng S (August 2008). "Simple multiplex RT-PCR for identifying common fusion transcripts in childhood acute leukemia". International Journal of Laboratory Hematology. 30 (4): 286–91. doi:10.1111/j.1751-553X.2007.00954.x. PMID 18665825.
- McWhirter JR, Neuteboom ST, Wancewicz EV, Monia BP, Downing JR, Murre C (September 1999). "Oncogenic homeodomain transcription factor E2A-Pbx1 activates a novel WNT gene in pre-B acute lymphoblastoid leukemia". Proceedings of the National Academy of Sciences of the United States of America. 96 (20): 11464–9. Bibcode:1999PNAS...9611464M. doi:10.1073/pnas.96.20.11464. PMC 18056. PMID 10500199.
- Rudolph C, Hegazy AN, von Neuhoff N, Steinemann D, Schröck E, Stripecke R, et al. (August 2005). "Cytogenetic characterization of a BCR-ABL transduced mouse cell line". Cancer Genetics and Cytogenetics. 161 (1): 51–6. doi:10.1016/j.cancergencyto.2004.12.021. PMID 16080957.
- Caslini C, Serna A, Rossi V, Introna M, Biondi A (June 2004). "Modulation of cell cycle by graded expression of MLL-AF4 fusion oncoprotein". Leukemia. 18 (6): 1064–71. doi:10.1038/sj.leu.2403321. PMID 14990976.
- Martín-Subero JI, Odero MD, Hernandez R, Cigudosa JC, Agirre X, Saez B, et al. (August 2005). "Amplification of IGH/MYC fusion in clinically aggressive IGH/BCL2-positive germinal center B-cell lymphomas". Genes, Chromosomes & Cancer. 43 (4): 414–23. doi:10.1002/gcc.20187. PMID 15852472. S2CID 2025900.
- Zalcberg IQ, Silva ML, Abdelhay E, Tabak DG, Ornellas MH, Simões FV, et al. (October 1995). "Translocation 11;14 in three children with acute lymphoblastic leukemia of T-cell origin". Cancer Genetics and Cytogenetics. 84 (1): 32–8. doi:10.1016/0165-4608(95)00062-3. PMID 7497440.
- Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, Sultan C (August 1976). "Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group". British Journal of Haematology. 33 (4): 451–8. doi:10.1111/j.1365-2141.1976.tb03563.x. PMID 188440. S2CID 9985915.
- "ACS :: How Is Acute Lymphocytic Leukemia Classified?". Archived from the original on 23 March 2008.
- DeAngelo DJ, Pui C. Acute lymphoblastic leukemia and lymphoblastic lymphoma. Chapter 19 of American Society of Hematology Self-Assessment Program. 2013. ISBN 9780982843512
- ^ Orkin SH, Nathan DG, Ginsburg D, et al. (2014). Nathan and Oski's Hematology and Oncology of Infancy and Childhood (8th ed.). Saunders. ISBN 978-1-4557-5414-4.
- Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. (May 2016). "The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia". Blood. 127 (20): 2391–405. doi:10.1182/blood-2016-03-643544. PMID 27069254. S2CID 18338178.
- Kyriakidis I, Kyriakidis K, Tsezou A (August 2022). "MicroRNAs and the Diagnosis of Childhood Acute Lymphoblastic Leukemia: Systematic Review, Meta-Analysis and Re-Analysis with Novel Small RNA-Seq Tools". Cancers. 14 (16): 3976. doi:10.3390/cancers14163976.
{{cite journal}}: CS1 maint: unflagged free DOI (link) - "Acute lymphoblastic leukemia (ALL) Information – Mount Sinai – New York". Mount Sinai Health System. Archived from the original on 3 August 2016. Retrieved 18 November 2017.
- ^ Hoffbrand V, Moss P, Pettit J (31 October 2006). Essential Haematology. Wiley. pp. 192–196. ISBN 978-1-4051-3649-5. Archived from the original on 21 March 2015. Retrieved 14 September 2013.
- ^ "Adult Acute Lymphoblastic Leukemia Treatment". National Cancer Institute. Retrieved 6 December 2017.
- Jabbour E, Thomas D, Cortes J, Kantarjian HM, O'Brien S (May 2010). "Central nervous system prophylaxis in adults with acute lymphoblastic leukemia: current and emerging therapies". Cancer. 116 (10): 2290–300. doi:10.1002/cncr.25008. PMID 20209620. S2CID 13634096.
- Yanada M (June 2015). "Time to tune the treatment of Ph+ ALL". Blood. 125 (24): 3674–5. doi:10.1182/blood-2015-04-641704. PMID 26069331.
- Seiter K, Harris JE. Acute Lymphoblastic Leukemia Treatment Protocols. emedicine; Medscape. "Archived copy". Archived from the original on 1 September 2015. Retrieved 16 August 2015.
{{cite web}}: CS1 maint: archived copy as title (link) - ^ Hoffbrand AV, Moss PA (26 October 2015). Hoffbrand's essential haematology (Seventh ed.). Chichester, West Sussex. ISBN 9781118408674. OCLC 909538759.
{{cite book}}: CS1 maint: location missing publisher (link) - Lambrou GI, Papadimitriou L, Chrousos GP, Vlahopoulos SA (April 2012). "Glucocorticoid and proteasome inhibitor impact on the leukemic lymphoblast: multiple, diverse signals converging on a few key downstream regulators". Molecular and Cellular Endocrinology. 351 (2): 142–51. doi:10.1016/j.mce.2012.01.003. PMID 22273806. S2CID 28749125.
- Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. (April 2013). "Chimeric antigen receptor-modified T cells for acute lymphoid leukemia". The New England Journal of Medicine. 368 (16): 1509–1518. doi:10.1056/NEJMoa1215134. PMC 4058440. PMID 23527958.
- ^ Barrett DM, Singh N, Porter DL, Grupp SA, June CH (2014). "Chimeric antigen receptor therapy for cancer". Annual Review of Medicine. 65: 333–47. doi:10.1146/annurev-med-060512-150254. PMC 4120077. PMID 24274181.
- Alonso-Camino V, Sánchez-Martín D, Compte M, Nuñez-Prado N, Diaz RM, Vile R, Alvarez-Vallina L (May 2013). "CARbodies: Human Antibodies Against Cell Surface Tumor Antigens Selected From Repertoires Displayed on T Cell Chimeric Antigen Receptors". Molecular Therapy: Nucleic Acids. 2: e93. doi:10.1038/mtna.2013.19. PMC 4817937. PMID 23695536.
- Zufferey R, Dull T, Mandel RJ, Bukovsky A, Quiroz D, Naldini L, Trono D (December 1998). "Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery". Journal of Virology. 72 (12): 9873–80. doi:10.1128/JVI.72.12.9873-9880.1998. PMC 110499. PMID 9811723.
- Office of the Commissioner. "Press Announcements—FDA approval brings first gene therapy to the United States". www.fda.gov. Archived from the original on 3 September 2017. Retrieved 12 September 2017.
- Ledford H (July 2017). "Engineered cell therapy for cancer gets thumbs up from FDA advisers". Nature. 547 (7663): 270. Bibcode:2017Natur.547..270L. doi:10.1038/nature.2017.22304. PMID 28726836.
- Kantarjian H, Stein A, Gökbuget N, Fielding AK, Schuh AC, Ribera JM, et al. (March 2017). "Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia". The New England Journal of Medicine. 376 (9): 836–847. doi:10.1056/nejmoa1609783. PMC 5881572. PMID 28249141.
- Estcourt L, Stanworth S, Doree C, Hopewell S, Murphy MF, Tinmouth A, Heddle N (May 2012). Cochrane Haematological Malignancies Group (ed.). "Prophylactic platelet transfusion for prevention of bleeding in patients with haematological disorders after chemotherapy and stem cell transplantation". The Cochrane Database of Systematic Reviews (5): CD004269. doi:10.1002/14651858.CD004269.pub3. PMID 22592695.
- Estcourt LJ, Stanworth SJ, Doree C, Hopewell S, Trivella M, Murphy MF (November 2015). Cochrane Haematological Malignancies Group (ed.). "Comparison of different platelet count thresholds to guide administration of prophylactic platelet transfusion for preventing bleeding in people with haematological disorders after myelosuppressive chemotherapy or stem cell transplantation". The Cochrane Database of Systematic Reviews. 2015 (11): CD010983. doi:10.1002/14651858.CD010983.pub2. PMC 4717525. PMID 26576687.
- Fisher SA, Cutler A, Doree C, Brunskill SJ, Stanworth SJ, Navarrete C, Girdlestone J (January 2019). Cochrane Haematological Malignancies Group (ed.). "Mesenchymal stromal cells as treatment or prophylaxis for acute or chronic graft-versus-host disease in haematopoietic stem cell transplant (HSCT) recipients with a haematological condition". The Cochrane Database of Systematic Reviews. 1: CD009768. doi:10.1002/14651858.CD009768.pub2. PMC 6353308. PMID 30697701.
- Knips L, Bergenthal N, Streckmann F, Monsef I, Elter T, Skoetz N (January 2019). Cochrane Haematological Malignancies Group (ed.). "Aerobic physical exercise for adult patients with haematological malignancies". The Cochrane Database of Systematic Reviews. 1: CD009075. doi:10.1002/14651858.CD009075.pub3. PMC 6354325. PMID 30702150.
- "FDA approves brexucabtagene autoleucel for relapsed or refractory B-cell precursor acute lymphoblastic leukemia". U.S. Food and Drug Administration (FDA). 1 October 2021. Retrieved 2 October 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain.
- "U.S. FDA Approves Kite's Tecartus as the First and Only Car T for Adults With Relapsed or Refractory B-cell Acute Lymphoblastic Leukemia". Kite. 1 October 2021. Retrieved 1 October 2021 – via Business Wire.
- ^ "FDA Approves First Cell-Based Gene Therapy For Adult Patients with Relapsed or Refractory MCL". U.S. Food and Drug Administration (FDA). 24 July 2020. Retrieved 24 July 2020. This article incorporates text from this source, which is in the public domain.
- Hutter JJ (June 2010). "Childhood leukemia". Pediatrics in Review. 31 (6): 234–41. doi:10.1542/pir.31-6-234. PMID 20516235.
- "Prognosis and survival for acute lymphocytic leukemia - Canadian Cance". www.cancer.ca. Retrieved 6 December 2017.
- Nelson Essentials of Pediatrics By Karen Marcdante, Robert M. Kliegman, Richard E. Behrman, Hal B. Jenson p597
- The Guide Paediatrics. ISBN 978-978-917-9909. p51
- Hoffbrand AV, Moss PA (26 October 2015). Hoffbrand's essential haematology (Seventh ed.). Chichester, West Sussex. p. 194. ISBN 9781118408674. OCLC 909538759.
{{cite book}}: CS1 maint: location missing publisher (link) - Moorman AV, Harrison CJ, Buck GA, Richards SM, Secker-Walker LM, Martineau M, et al. (April 2007). "Karyotype is an independent prognostic factor in adult acute lymphoblastic leukemia (ALL): analysis of cytogenetic data from patients treated on the Medical Research Council (MRC) UKALLXII/Eastern Cooperative Oncology Group (ECOG) 2993 trial". Blood. 109 (8): 3189–97. doi:10.1182/blood-2006-10-051912. PMID 17170120. S2CID 1038016.
- Den Boer ML, van Slegtenhorst M, De Menezes RX, Cheok MH, Buijs-Gladdines JG, Peters ST, et al. (February 2009). "A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study". The Lancet. Oncology. 10 (2): 125–34. doi:10.1016/S1470-2045(08)70339-5. PMC 2707020. PMID 19138562.
- Guo LM, Xi JS, Ma Y, Shao L, Nie CL, Wang GJ (January 2014). "ARID5B gene rs10821936 polymorphism is associated with childhood acute lymphoblastic leukemia: a meta-analysis based on 39,116 subjects". Tumour Biology. 35 (1): 709–13. doi:10.1007/s13277-013-1097-0. PMID 23975371. S2CID 12601034.
- Greer JP, Arber DA, Glader B, et al. (2013). Wintrobe's Clinical Hematology (13th ed.). Lippincott Williams & Wilkins. ISBN 978-1-4511-7268-3.
- Urayama KY, Manabe A (October 2014). "Genomic evaluations of childhood acute lymphoblastic leukemia susceptibility across race/ethnicities". the Japanese Journal of Clinical Hematology. 55 (10): 2242–8. PMID 25297793.
- Ries LA, Smith MA, Gurney JG, Linet M, Tamra T, Young JL, Bunin GR (1999). Cancer Incidence and Survival among Children and Adolescents: United States SEER Program 1975-1995. Bethesda, MD: National Cancer Institute, SEER Program.
- ^ Shapira T, Pereg D, Lishner M (September 2008). "How I treat acute and chronic leukemia in pregnancy". Blood Reviews. 22 (5): 247–59. doi:10.1016/j.blre.2008.03.006. PMID 18472198.
External links
- Acute Lymphocytic Leukemia Archived 16 November 2009 at the Wayback Machine at the American Cancer Society
- Childhood ALL Treatment at the National Cancer Institute
| Classification | D |
|---|---|
| External resources |
| Leukaemias, lymphomas and related disease | |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||
| Cutaneous lymphoid hyperplasia | |||||||||||||||||||||||||||||||||||||||||||||
| General | |||||||||||||||||||||||||||||||||||||||||||||